
Structure, function, and regulation of mammalian TRPC channels, mechanisms of Ca2+ dyshomeostasis during oxidative stress
The long range goal of my research is to understand the molecular mechanisms associated with agonist-induced Ca2+ signaling in mammalian non-excitable cells and in particular, how Ca2+ signaling may be altered during oxidative stress conditions. Our major focus is on understanding Ca2+ homeostasis in vascular endothelial cells and renal epithelial cells. TRP genes, originally identified as critical components of phototransduction in Drosophila, encode a ubiquitous and heterogeneous family of ion channel proteins that play a fundamental role in Ca2+ signaling. There are currently 28 mammalian homologs divided among 6 subgroups. The seven mammalian TRP siblings designated TRPC1-TRPC7, exhibit ~38% overall identity at the amino acid level to the Drosophila isoforms. The primary TRPC channels, like the Drosophila versions, are regulated by phospholipase C-dependent mechanisms and are thus, intimately involved in receptor-mediated Ca2+ signaling.
Ca2+ Tunneling Hypothesis for Ca2+ Reabsorption in Principal Cells of the Collecting Duct
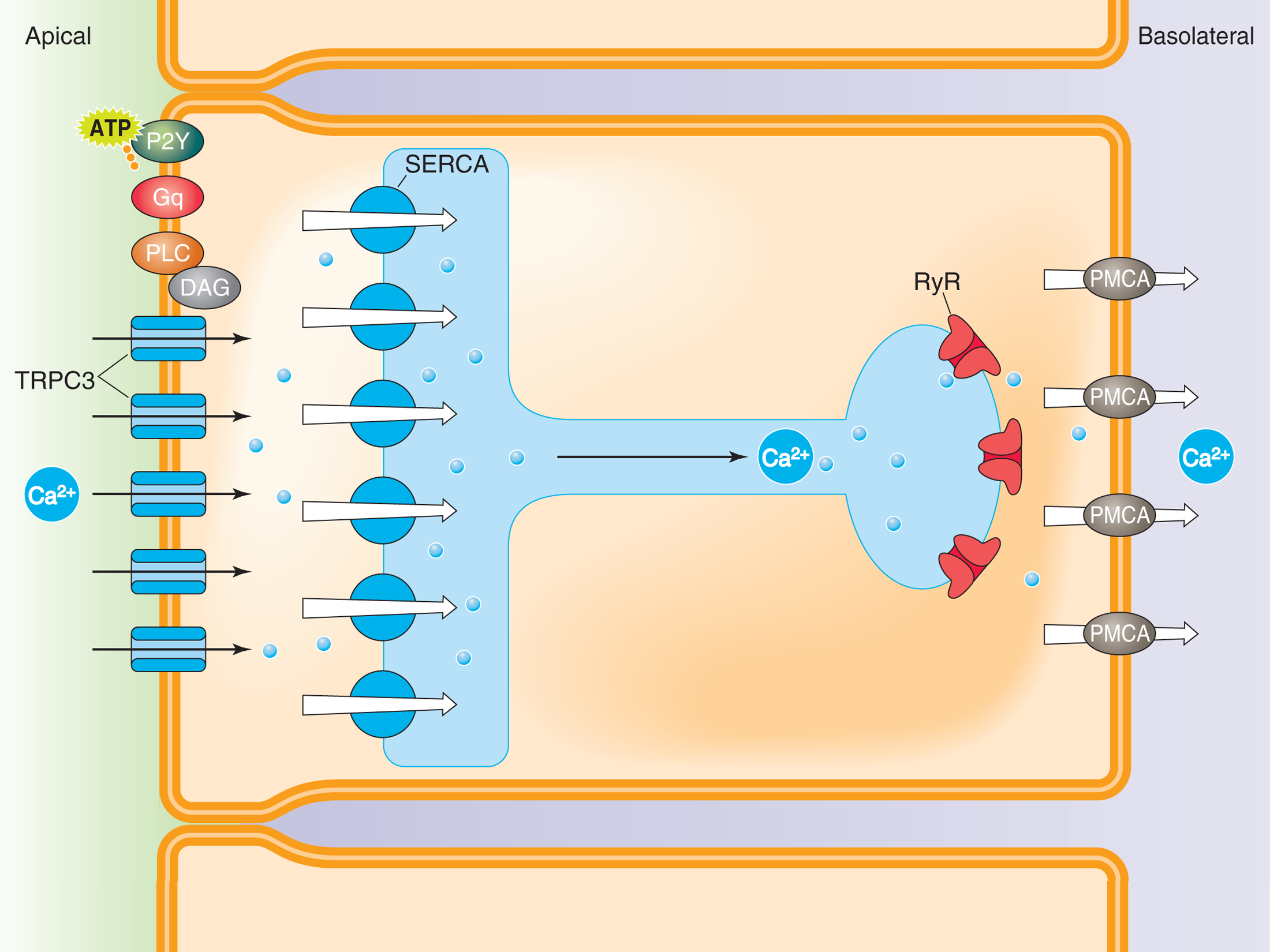
Ca2+ bypasses the global cytoplasm by “tunneling” to the basolateral domain via the endoplasmic reticulum. Ca2+ 1) enters the cell at the apical membrane via TRPC3 channels, 2) is immediately pumped into the endoplasmic reticulum (ER) by the sarcoplasmic-endoplasmic reticulum Ca2+-ATPase (SERCA) pump, 3) is translocated from the apical to the basolateral domains of the cell via the ER, 4) is released into the cytoplasm via ryanodine receptors (RyR), and 5) is ultimately pumped out of the cell at the basolateral membrane by the plasmalemmal Ca2+-ATPase (PMCA) pump.
The Potential Role of TRPC3 Channels in Kidney Stone Formation
Kidney stone formation is one of the most common ailments of the urinary track in the United States today and a growing health concern. A major risk factor for the development of stone disease is dehydration. The antidiuretic hormone, arginine-vasopressin (AVP) controls water homeostasis and urine concentration by stimulating the translocation of the water channel, aquaporin-2 (AQP2) from intracellular vesicles to the apical membrane of principal cells of the collecting duct (CD) where it dramatically increases water permeability. Since urinary Ca2+ concentration ([Ca2+]U) plays a critical role in stone formation, there must be endogenous physiological mechanisms that allow the kidney to conserve water during dehydration without causing a dangerous rise in [Ca2+]U. In this regard, AVP also increases Ca2+ reabsorption in the distal nephron, but the mechanism(s) by which this occurs is unclear.
Our recent studies have shown that the transient receptor potential channel, TRPC3 co-localizes with AQP2 in principal cells of the CD and accumulates in the apical membrane in response to AVP. TRPC3 is a Ca2+ permeable, non-selective cation channel activated downstream of phospholipase C. Once in the apical membrane, we found that TRPC3 is essential for net apical-to-basolateral transepithelial Ca2+ flux across high resistance CD cell monolayers, but the role of TRPC3 in Ca2+ reabsorption during antiduresis in vivo, is unclear. Additionally, the role of other transport proteins involved in transcellular Ca2+ movement, and the functional consequences of co-regulation of TRPC3 and AQP2 trafficking by AVP remains unknown. Our overall hypothesis is that AVP-induced Ca2+ reabsorption via TRPC3 in the CD reflects a fundamental mechanism that has evolved to allow dramatic water reabsorption during periods of dehydration while limiting the rise in tubular Ca2+ and thus, limiting the risk of nephrocalcinosis and urolithiasis, i.e., kidney stone disease. In this project, we will use mice with a collecting-duct specific knockout of TRPC3 to test the hypothesis that TRPC3 channels control the final urinary Ca2+ concentration in response to changes in water balance. We will use fluorescent Ca2+ imaging techniques and radiotracer flux in isolated perfused tubules to trace the molecular pathway of vectorial Ca2+ transport by principal cells of the CD. And we will combine molecular biological and biochemical techniques with functional studies to test the hypothesis that AQP2 water channels regulate Ca2+ reabsorption by physical and functional interaction with TRPC3 channels. Overall, our experiments will test an entirely new hypothesis for how water is reabsorbed in response to AVP during periods of dehydration without causing a rise in urinary Ca2+. These studies will help identify novel molecular targets for the ultimate prevention of urinary stone disease.
Regulation of the inositol 1,4,5 trisphosphate receptor by glutathionylation
Ca2+ signaling plays an essential role in the integrative function of the vascular endothelium. Receptor-initiated changes in cytosolic free Ca2+ concentration ([Ca2+]i) of the endothelial cell (EC) trigger the production and release of paracrine factors that control vasoreactivity and permeability, inflammation, hemostasis and angiogenesis. Because of their unique location, ECs are susceptible to a diverse range of oxidative insults. One of the earliest detectable cellular events associated with oxidative stress is an increase in basal [Ca2+]i and a dysruption in normal Ca2+ signaling, but the molecular mechanisms by which this occurs are unclear. Oxidative stress is defined as an imbalance between the amount of reactive oxygen species and the normal antioxidant defenses of the cell leading to changes in molecular signaling or control. The primary antioxidant of the cell is glutathione (GSH). Protein S-glutathionylation (GS-ylation), the reversible post-translational modification of sensitive cysteine (Cys) residues by GSH, regulates protein function during physiological redox signaling and may contribute to detrimental changes in protein structure and function during oxidative stress.
We recently discovered that the inositol 1,4,5-trisphosphate receptor (IP3R) in vascular endothelial cells (ECs) is GS-ylated in response to both physiological and pharmacological oxidants. Modification of the IP3R by GS-ylation correlates with increased spontaneous Ca2+ oscillations and enhanced cell sensitivity to vasoactive agonists, bradykinin and histamine. The overall goal of this project is to understand the molecular mechanisms by which GS-ylation relates IP3R function during redox signaling and during oxidative stress. In this project, we will test the hypothesis that GS-ylation increases sensitivity of the IP3R to IP3, i.e., enhanced IP3–induced Ca2+ release; and we will test the alternative hypothesis that GS-ylation increases sensitivity of the IP3R to Ca2+, i.e., enhanced Ca2+-induced Ca2+ release. We will determine which Cys residues of the IP3R are modified by GS-ylation and which are necessary for changes in IP3R channel activity. We will also test the hypothesis that ERp44, an member of the thioredoxin family of proteins that is localized to the ER lumen, catalyzes IP3R GS-ylation via disulfide exchange reaction, and we will determine if GS-ylation is a molecular switch linking oxidative protein folding to IP3R function. Lastly we will test the hypothesis that the IP3R is GS-ylated in vivo during development of metabolic syndrome, and determine if this modification is responsible for enhance sensitivity of platelets observed. This study will provide an integrative understanding of how cellular oxidants regulate IP3R function in vascular ECs under both physiological and pathological conditions.
The role of PMCA pump-channels in oxidative stress-induced Ca2+ overload
Ion channels use the energy stored in ionic gradients to initiate rapid signaling events essential for the function of virtually every cell in the body. Although channel activation can occur in response to a variety of different stimuli, once open all channels share a common feature—they allow millions of ions per second to cross the membrane. Pump proteins on the other hand, use the energy in ATP to establish the ionic gradients necessary for channel function. Pumps generally move hundreds of ions per second, and hence their density in the membrane is by necessity much higher than that of channels. These striking differences led to the view that pumps and channels move ions by very different mechanisms. However, studies on the potent marine toxin, palytoxin (PTX) challenge this concept. PTX binds with picomolar affinity to the Na+,K+-ATPase (NKA) and converts the pump into a non-selection cation channel. Evaluation of PTX action suggests that the fundamental difference between channels and pumps resides not so much in the molecular architecture of the ion translocation pathway itself, by rather in the intrinsic gating properties of the protein. More importantly, the fact that high affinity toxins can induce a channel-mode of pump operation suggests that endogenous mechanism may also exist that lead to the same channel mode and which may have either an important physiological role in cell signaling, or produce disastrous consequences for cell function and survival if not adequately controlled.
Our studies have shown that another marine toxin called maitotoxin (MTX), converts the plasmalemmal Ca2+-ATPase pump (PMCA) into a Ca2+ permeable, non-selective cation channel which ultimately causes Ca2+ overload-induced necrotic cell death. Furthermore, we discovered that the Ca2+ channels activated by MTX in vascular endothelial cells are biophysically identical to the channels activated by the model oxidant, tert-butyl-hydroperoxide or by oxidized glutathione GSSG). Thus, changes in the redox status of the cell may, like MTX, trigger conversion of the PMCA pump into a channel. In this project, we will test the hypothesis that glutathionylation not only inhibits PMCA pump activity, but actually converts the PMCA into a channel much like MTX. We will determine if the PMCA can be directly glutathionylated in vivo during oxidative stress and determine if the PMCA pump-to-channel conversion plays a significant role in altered Ca2+ homeostasis. The conversion of the PMCA pump into a channel provides a novel and ubiquitous mechanism by which oxidative stress initiates Ca2+-overload and provides a new molecular target for therapeutic intervention in a variety of pathological conditions associated with oxidative stress, including atherosclerosis, ischemia-reperfusion injury, Alzheimer’s and Parkinson’s disease, and perhaps biological aging.
| Major Research Areas |
|---|
| Ion Channels, Membrane Transport, Protein Structure/Function |
| Disciplines |
| Biophysics, Electrophysiology, Fluorescence Spectroscopy, Signal Transduction |
| Organ Systems |
| Cardiovascular System, Nervous System, Renal System |
| Diseases |
| Channelopathies, Kidney Stones |
| Major Techniques |
| Cell Culture, Cellular and Molecular Biology, Confocal Microscopy, Electrophysiology, Gel Electrophoresis/Western Blots, Immunohistochemistry, In-vivo Animal Models, Ion Transport, Mass Spectrometry, Patch Clamping, Protein Expression, Protein-Protein Interactions (Biocore), Quantitative Fluorescence Imaging, Working Heart Perfusions |
| Genes |
| TRPC3 (), TRPC6 () |
